
Joanna Le Noury, psicologa ricercatrice1, John M. Nardo 2 , professore assistente clinico in pensione 2, David Healy , professore 1, Jon Jureidini, professore clinico 3 , Melissa Raven , borsista post-dottorato 3 , Catalin Tufanaru, ricercatore associato 4, Elia Abi-Jaoude, psichiatra del personale 5
Pubblicato su BMJ,2015
Traduzione in italiano a cura del Dott. Claudio Ajmone per GiùleManidaiBambini.org
Testo originale in inglese, disponibile a questo link
- School of Medical Sciences, Bangor University, Bangor, Wales, UK
- Emory University, Atlanta, Georgia, USA
- Critical and Ethical Mental Health Research Group, Robinson Research Institute, University of Adelaide, Adelaide, South Australia, Australia
- Joanna Briggs Institute, Faculty of Health Sciences, University of Adelaide, Adelaide, South Australia, Australia
- Department of Psychiatry, The Hospital for Sick Children, University of Toronto, Toronto, Ontario, Canada
Riassunto
Obiettivi: Rianalizzare lo studio 329 di SmithKline Beecham (pubblicato da Keller e colleghi nel 2001), il cui obiettivo primario era confrontare l’efficacia e la sicurezza di paroxetina e imipramina con placebo nel trattamento di adolescenti con depressione maggiore unipolare. La rianalisi nell’ambito dell’iniziativa RIAT (ripristino degli studi invisibili e abbandonati) è stata eseguita per verificare se l’accesso e la rianalisi di un set di dati completo da uno studio controllato randomizzato avrebbero implicazioni clinicamente rilevanti per la medicina basata sull’evidenza.
Progettazione: Studio in doppio cieco randomizzato controllato con placebo.
Contesto: 12 centri psichiatrici accademici nordamericani, dal 20 aprile 1994 al 15 febbraio 1998.
Partecipanti: 275 adolescenti con depressione maggiore della durata di almeno otto settimane. I criteri di esclusione includevano una serie di disturbi psichiatrici e medici concomitanti e il suicidio.
Interventi: I partecipanti sono stati randomizzati a otto settimane di trattamento in doppio cieco con paroxetina (20-40 mg), imipramina (200-300 mg) o placebo.
Principali misure di esito: Le variabili di efficacia primarie prespecificate erano il cambiamento dal basale alla fine della fase di trattamento acuto di otto settimane nel punteggio totale della scala della depressione di Hamilton (HAM-D) e la percentuale di responder (punteggio HAM-D ≤8 o riduzione ≥50% nell’HAM-D basale) all’endpoint acuto. Gli esiti secondari prespecificati erano cambiamenti dal basale all’endpoint negli item relativi alla depressione nel K-SADS-L, impressione clinica globale, lista di controllo del funzionamento autonomo, profilo di auto-percezione e scala dell’impatto della malattia; predittori di risposta; e il numero di pazienti che ricadono durante la fase di mantenimento. Le esperienze avverse dovevano essere confrontate principalmente utilizzando statistiche descrittive. Nessun dizionario di codifica è stato prespecificato.
Risultati: L’efficacia di paroxetina e imipramina non era statisticamente o clinicamente significativamente diversa dal placebo per qualsiasi risultato di efficacia primario o secondario prespecificato. I punteggi HAM-D sono diminuiti di 10,7 (media dei minimi quadrati) (intervallo di confidenza al 95% da 9,1 a 12,3), 9,0 (da 7,4 a 10,5) e 9,1 (da 7,5 a 10,7) punti, rispettivamente, per i gruppi paroxetina, imipramina e placebo (P = 0,20). Ci sono stati aumenti clinicamente significativi dei danni, inclusi ideazione e comportamento suicidari e altri eventi avversi gravi nel gruppo paroxetina e problemi cardiovascolari nel gruppo imipramina.
Conclusioni: Né la paroxetina né l’imipramina ad alte dosi hanno mostrato efficacia per la depressione maggiore negli adolescenti e c’è stato un aumento dei danni con entrambi i farmaci. L’accesso ai dati primari degli studi ha importanti implicazioni sia per la pratica clinica che per la ricerca, compreso il fatto che le conclusioni pubblicate sull’efficacia e la sicurezza non dovrebbero essere lette come autorevoli. La nuova analisi dello Studio 329 illustra la necessità di rendere disponibili i dati e i protocolli degli studi primari per aumentare il rigore della base di prove.
Introduzione
Nel 2013, di fronte alla segnalazione selettiva dei risultati di studi randomizzati controllati, un gruppo internazionale di ricercatori ha invitato finanziatori e ricercatori di studi abbandonati (non pubblicati) o riportati in modo errato per pubblicare risultati non divulgati o correggere pubblicazioni fuorvianti. 1 Questa iniziativa è stata chiamata “ripristino di processi invisibili e abbandonati” (RIAT). I ricercatori hanno identificato molte prove che richiedono una revisione e hanno inviato un’e-mail ai finanziatori, chiedendo loro di segnalare la loro intenzione di pubblicare le prove non pubblicate o pubblicare versioni corrette di prove riportate in modo errato. Se finanziatori e ricercatori non si sono impegnati a correggere uno studio che era stato identificato come non pubblicato o riportato in modo errato, i gruppi indipendenti sono stati incoraggiati a pubblicare una rappresentazione accurata della sperimentazione clinica sulla base delle informazioni normative pertinenti.
L’articolo attuale rappresenta una pubblicazione RIAT dello studio 329. Lo studio originale è stato finanziato da SmithKline Beecham (SKB; successivamente GlaxoSmithKline, GSK). Riconosciamo il lavoro degli investigatori originali. Questo studio controllato randomizzato in doppio cieco per valutare l’efficacia e la sicurezza di paroxetina e imipramina rispetto al placebo per adolescenti con diagnosi di depressione maggiore è stato riportato nel Journal of the American Academy of Child and Adolescent Psychiatry (JAACAP) nel 2001, con Martin Keller come il autore principale. 2
I ricercatori del RIAT hanno identificato lo Studio 329 come un esempio di uno studio segnalato in modo errato che necessita di revisione. L’articolo di Keller e colleghi, in gran parte scritto da fantasmi, 3 ha dichiarato l’efficacia e la sicurezza per la paroxetina che era in contrasto con i dati. 4 Questo è problematico perché l’articolo è stato influente nella letteratura a sostegno dell’uso di antidepressivi negli adolescenti. 5
Il 14 giugno 2013, i ricercatori del RIAT hanno chiesto a GSK se avesse intenzione di rivedere uno qualsiasi degli studi sponsorizzati, incluso lo studio 329. GSK non ha segnalato alcuna intenzione di pubblicare una versione corretta di nessuno dei suoi studi. In una successiva corrispondenza, GSK ha affermato che lo studio di Keller e colleghi “riflette accuratamente le opinioni oneste degli autori degli investigatori clinici” e che GSK “non era d’accordo sul fatto che l’articolo è falso, fraudolento o fuorviante”. 6
Lo studio 329 era uno studio multicentrico controllato randomizzato in doppio cieco di otto settimane (fase acuta), seguito da una fase di continuazione di sei mesi. L’obiettivo primario dichiarato di SKB era quello di esaminare l’efficacia e la sicurezza di imipramina e paroxetina rispetto al placebo nel trattamento di adolescenti con depressione maggiore unipolare. Gli obiettivi secondari erano identificare i predittori degli esiti del trattamento attraverso i sottotipi clinici; fornire informazioni sul profilo di sicurezza di paroxetina e imipramina quando questi farmaci sono stati somministrati per “un periodo di tempo prolungato”; e per stimare il tasso di ricaduta tra i pazienti che hanno risposto a imipramina, paroxetina e placebo e sono stati mantenuti durante il trattamento. L’iscrizione allo studio è avvenuta tra l’aprile 1994 e il marzo 1997.
La prima pubblicazione dello studio RIAT era uno studio chirurgico che era stato pubblicato solo in parte prima. 7 Pochi studi clinici controllati randomizzati pubblicati in precedenza sono stati riportati successivamente in articoli pubblicati da diversi gruppi di autori.8
Metodi
Abbiamo rianalizzato i dati dello studio 329 secondo le raccomandazioni del RIAT. A tal fine, abbiamo utilizzato il rapporto dello studio clinico (il “rapporto clinico finale” di SKB), comprese le appendici AG, che sono disponibili pubblicamente sul sito Web GSK, altri9 documenti disponibili pubblicamente,10 e i dati dei singoli partecipanti accessibili tramite il sito Web di SAS Solutions OnDemand, 11 su cui GSK ha successivamente pubblicato anche alcuni documenti dello studio 329 (disponibili solo per gli utenti approvati da GSK). Dopo la negoziazione,12 GSK ha pubblicato circa 77.000 pagine di moduli di segnalazione di casi individuali non identificati (appendice H) su quel sito web.
Abbiamo utilizzato uno strumento per documentare la trasformazione da documenti normativi a pubblicazione su rivista, sulla base della checklist di informazioni CONSORT 2010 da includere quando si riporta uno studio randomizzato. La registrazione dell’audit, inclusa una tabella delle fonti di dati consultate nella preparazione di ciascuna parte di questo documento, è disponibile nell’appendice 1. Tranne dove indicato, in conformità con le raccomandazioni RIAT, i nostri metodi sono quelli stabiliti nel protocollo 1994-96 per lo studio 329. 13 Nei casi in cui i metodi utilizzati e pubblicati da Keller e colleghi divergessero dal protocollo, abbiamo seguito il protocollo originale.
Poiché il protocollo ha specificato il metodo di correzione per i valori mancanti – ultima osservazione portata avanti – è stato messo in discussione negli anni successivi, abbiamo incluso anche un metodo più moderno – imputazione multipla – su richiesta deirevisori tra pari del BM. Questo è un metodo post hoc aggiunto solo per confronto e non fa parte della nostra rianalisi formale. Quando il protocollo non era specifico, abbiamo scelto per consenso i metodi standard che presentavano al meglio i dati. Il protocollo originale del 1993 aveva emendamenti minori nel 1994 e nel 1996 (sostituzione del Shedule for Affettcyiv Disorder and Schizophrenia for Adolescente-Present Version con la Lifetime Version (K-SADS-L) e riduzione della dimensione del campione richiesta). Inoltre, il rapporto dello studio clinico (CSR) riportava alcune procedure diverse da quelle specificate nel protocollo. Abbiamo notato variazioni che abbiamo ritenuto rilevanti.
Partecipanti
Lo studio originale ha reclutato 275 adolescenti di età compresa tra 12 e 18 anni che soddisfacevano i criteri del DSM-IV 14 per un episodio in corso di depressione maggiore della durata di almeno otto settimane (il protocollo specificava i criteri DSM-III-R, che sono simili). Il riquadro 1 elenca i criteri di ammissibilità.
Riquadro 1: criteri di ammissibilità dello studio
Criterio di inclusione
- Adolescenti di età compresa tra 12 e 18 anni che hanno soddisfatto i criteri del DSM-III-R per la depressione maggiore per almeno 8 settimane
- Punteggio di gravità <60 sulla scala di valutazione globale dei bambini (CGAS)
- Punteggio ≥12 sulla scala della depressione di Hamilton (17 item) (HAM-D)
- Salute dal punto di vista medico
- QI ≥80 (basato sul test del vocabolario illustrato di Peabody)
Criteri di esclusione
- Diagnosi DSM-III-R attuale o passata di disturbo bipolare, disturbo schizoaffettivo, anoressia nervosa, bulimia, abuso / dipendenza da alcol o droghe, disturbo ossessivo-compulsivo, autismo/ disturbo mentale pervasivo o disturbo psichiatrico organico
- Diagnosi attuale (entro 12 mesi) DSM-III-R di disturbo da stress post-traumatico
- Prova adeguata di un antidepressivo entro sei mesi (trattamento di almeno quattro settimane con una dose adeguata di antidepressivo)
- Ideazione suicidaria con un piano preciso, tentativo di suicidio durante l’attuale episodio depressivo o anamnesi di tentato suicidio per overdose di droga
- Malattia medica che controindica l’uso di antidepressivi eterociclici
- Uso corrente di farmaci psicotropi (inclusi ansiolitici, antipsicotici, stabilizzatori dell’umore) o droghe illecite
- Malattia cerebrale organica, epilessia o “ritardo mentale”
- Gravidanza o allattamento
- Donne sessualmente attive che non usano una contraccezione affidabile
- Uso di un farmaco sperimentale nei 30 giorni precedenti o cinque emivite del farmaco sperimentale
Un numero sconosciuto di pazienti (non divulgato nei documenti disponibili) identificati tramite screening telefonico come potenziali partecipanti è stato successivamente valutato presso il sito dello studio da un medico senior (psichiatra o psicologo). La società sponsorizzatrice ha tenuto più riunioni e teleconferenze con i ricercatori dello studio del sito per garantire la standardizzazione tra i siti. Pazienti e genitori sono stati intervistati separatamente con il K-SADS-L. Dopo questa valutazione iniziale, il paziente e il genitore hanno firmato entrambi il modulo di consenso informato dello studio; non vi era alcuna menzione di un modulo di consenso separato nel protocollo o nella CSR. È stato utilizzato un periodo di screening da sette a dieci giorni per ottenere le cartelle cliniche precedenti e per documentare che i sintomi depressivi erano stabili. Alla fine del periodo di valutazione, solo i pazienti che continuavano a soddisfare i criteri di inclusione (depressione maggiore DSM-III-R e punteggio totale HAM-D ≥12) sono stati randomizzati. Non c’era alcuna fase di introduzione al placebo.
In origine c’erano sei siti di studio, ma questo numero è stato aumentato a 12 (10 negli Stati Uniti e due in Canada). I centri erano affiliati a un dipartimento di psichiatria universitaria o ospedaliera e avevano esperienza con pazienti adolescenti. I ricercatori sono stati selezionati in base al loro interesse per lo studio e alla loro capacità di reclutare i pazienti dello studio. Il periodo di reclutamento è andato dal 20 aprile 1994 al 15 marzo 1997 e la fase apicale è stata completata il 7 maggio 1997. In un piccolo numero di pazienti, i dati di follow-up a 30 giorni per i casi che sono entrati nella fase di continuazione sono stati raccolti nel febbraio 1998 .
Coinvolgimento del paziente
Per quanto possiamo accertare, non vi è stato alcun coinvolgimento dei pazienti nel disegno dello studio della SKB.
Interventi
Il farmaco in studio è stato fornito ai pazienti in blister settimanali. I pazienti sono stati istruiti a prendere il farmaco due volte al giorno. C’erano sei livelli di dosaggio. Durante le prime quattro settimane, tutti i pazienti sono stati titolati al livello quattro, corrispondente a 20 mg di paroxetina o 200 mg di imipramina, indipendentemente dalla risposta. I non responder (quelli che non riescono a raggiungere i criteri di responder) potrebbero essere titolati fino al livello cinque o sei nelle successive quattro settimane. Ciò corrisponde a dosi massime di 60 mg di paroxetina e 300 mg di imipramina.
La conformità al trattamento è stata valutata in base al numero di capsule erogate, prese e restituite. La non conformità è stata definita come l’assunzione di meno dell’80% o più del 120% del numero di capsule, valutata in base al numero previsto per essere restituito in due visite consecutive e ha comportato il ritiro. Anche i pazienti che mancavano di due visite consecutive sono stati ritirati dallo studio. Ai pazienti sono state fornite sessioni settimanali di 45 minuti di psicoterapia di supporto,15 principalmente allo scopo di valutare gli effetti del trattamento.
Dimensione del campione
La fase acuta dello studio era inizialmente basata su un’analisi di potenza che indicava che una dimensione del campione di 100 pazienti per gruppo di trattamento era necessaria per avere una potenza statistica dell’80% per un α a due code di 0,05 e una dimensione dell’effetto di 0,40. Questa dimensione dell’effetto ha comportato una differenza di 4 nel punteggio totale HAM-D dal basale all’endpoint, specificato nel protocollo per essere sufficientemente grande da essere clinicamente significativo, considerando una deviazione standard di 10. Non è stata fatta alcuna tolleranza nel calcolo della potenza per l’attrito (tasso di abbandono previsto) o non conformità durante lo studio.
Il reclutamento è stato più lento del previsto e, secondo quanto riferito, le scorte di trattamento (principalmente placebo) si sono esaurite a causa del superamento della data di scadenza. I ricercatori hanno effettuato una valutazione intermedia di 189 pazienti, senza rompere l’accecamento, che ha mostrato una minore variabilità nei punteggi HAM-D (SD 8) del previsto. Pertanto l’obiettivo di reclutamento è stato ridotto a 275 in quanto non avrebbe avuto un impatto negativo sulla potenza stimata dell’80% necessaria per rilevare una differenza di 4 punti tra placebo e gruppi di farmaci attivi.
Randomizzazione
Un elenco di randomizzazione generato dal computer di 360 numeri per la fase acuta è stato generato e tenuto da SKB. Secondo il CSR, i trattamenti sono stati bilanciati in blocchi di sei pazienti consecutivi; tuttavia, c’è un’incongruenza in quell’appendice. Un codice di randomizzazione descrive in dettaglio le dimensioni dei blocchi di sei e otto. A ogni ricercatore è stato assegnato un blocco di confezioni di trattamento numerate consecutivamente e ai pazienti sono stati assegnati numeri di trattamento in rigoroso ordine sequenziale. I pazienti sono stati randomizzati in un rapporto 1: 1: 1 al trattamento con paroxetina, imipramina o placebo.
Accecamento
La paroxetina è stata fornita sotto forma di compresse rivestite con film di colore giallo (10 mg) e rosa (20 mg) a forma di capsula. L’imipramina (50 mg) è stata acquistata in commercio e fornita sotto forma di compresse rotonde da 50 mg rivestite con film di colore verde. “Placebo paroxetina” corrispondeva alle compresse di paroxetina da 20 mg e “placebo imipramina” corrispondeva alle compresse di imipramina. Tutte le compresse sono state sovraincapsulate in capsule verde-bluastre per preservare l’accecamento. L’accecamento doveva essere interrotto solo in caso di un evento avverso grave che lo sperimentatore pensava non potesse essere adeguatamente trattato senza conoscere l’identità del trattamento in studio assegnato. L’identità del trattamento in studio non doveva essere altrimenti rivelata allo sperimentatore o al personale SKB associato allo studio.
Risultati
I pazienti sono stati valutati settimanalmente per le seguenti variabili di esito durante le otto settimane della fase di trattamento acuto.
Variabili primarie di efficacia
Le variabili di efficacia primarie prespecificate erano la variazione del punteggio totale su HAM-D 16 dall’inizio della fase di trattamento all’endpoint della fase acuta e la percentuale di responders alla fine della fase di trattamento acuto di otto settimane (più lunga di molti studi con antidepressivi). I responders sono stati definiti come pazienti che avevano una riduzione ≥50% dell’HAM-D o un punteggio HAM-D ≤8. (I punteggi sull’HAM-D possono variare da 0 a 52.)
Variabili secondarie di efficacia
Le variabili di efficacia secondarie prespecificate erano:
- Cambiamenti dalla linea basale all’endpoint in:
– Items depressione in K-SADS-L
-Impressione globale clinica (CGI)
-Lista di controllo per il funzionamento autonomo 17
-Profilo di percezione di sé
-Scala dell’impatto della malattia.
- Predittori di risposta (sottotipi endogeni, età, episodi precedenti, durata e gravità dell’episodio attuale, comorbilità con ansia separata, deficit di attenzione e disturbo della condotta)
- Il numero di pazienti che hanno avuto una recidiva durante la fase di mantenimento (indicata nella CSR e in questo documento come “fase di continuazione”).
Sia prima che dopo aver rotto il cieco, tuttavia, gli sponsor hanno apportato modifiche ai risultati secondari come descritto in precedenza. 4 Non siamo riusciti a trovare alcun documento che fornisse una motivazione scientifica per questi cambiamenti post hoc, 18 e pertanto i risultati non sono riportati in questo documento.
Sfide nello svolgimento di RIAT
A nostra conoscenza, questa è la prima analisi RIAT di uno studio segnalato in modo errato da un team esterno di autori, quindi non ci sono precedenti o guide chiare. Le sfide che abbiamo incontrato includevano:
Distorsione potenziale o percepita
Un rapporto RIAT non vuole essere una critica di una precedente pubblicazione. Il punto è piuttosto quello di produrre un’approfondita analisi indipendente di uno studio che è rimasto inedito o messo in discussione. Riconosciamo, tuttavia, che qualsiasi team RIAT potrebbe essere visto come un pregiudizio intrinseco in quanto mettere in discussione le conclusioni pubblicate in precedenza è ciò che ha riunito alcuni membri del team. Di conseguenza, abbiamo adottato tutte le misure procedurali appropriate per evitare tali presunti pregiudizi. Inoltre, abbiamo messo i dati a disposizione di altri per analizzarli.
Correzione per testare più variabili
Avevamo più fonti di informazione: il protocollo; il documento pubblicato; i documenti pubblicati sul sito web di GSK, inclusi la CSR e i dati dei singoli pazienti; e i dati primari grezzi nei moduli di segnalazione dei casi forniti da GSK su un desktop remoto per questo progetto. Il protocollo ha dichiarato due variabili primarie e sei variabili secondarie per i tre gruppi di trattamento in due diversi set di dati (caso osservato e ultima osservazione portata avanti). La CSR conteneva confronti statistici su 28 variabili discrete utilizzando due confronti (paroxetina vs placebo e imipramina vs placebo) nei due set di dati (caso osservato e ultima osservazione portata avanti). Il documento pubblicato elencava otto variabili con due confronti statistici ciascuno in un set di dati (ultima osservazione portata avanti). Gli autori dell’articolo originale, tuttavia, non hanno affrontato la necessità di correzioni per più variabili, un requisito standard quando ci sono più misure di risultato. In ultima analisi, non c’erano risultati statisticamente o clinicamente significativi per alcuna variabile di risultato, quindi non erano necessarie correzioni per questa analisi.
Test statistici
Il protocollo richiedeva il test ANOVA (modello lineare generalizzato) per variabili continue utilizzando un modello che includeva gli effetti del sito, del trattamento e dell’interazione sito × trattamento, con quest’ultimo caduto se P≥0.10. La regressione logistica (2 × 3 χ 2) è stata prescritta per le variabili categoriali nello stesso modello. Entrambi i metodi iniziano con una statistica omnibus per la significatività complessiva del set di dati, quindi passano al test a coppie se, e solo se, la statistica omnibus soddisfa α = 0,05. Tuttavia tutti i risultati statistici nel CSR e nel documento pubblicato sono stati riportati solo come valori a coppie solo per due dei tre possibili confronti (paroxetina vs placebo e imipramina vs placebo), senza menzionare la statistica omnibus. Pertanto, abbiamo condotto le analisi omnibus richieste, con risultati negativi come mostrato. I valori a coppie sono disponibili nella tabella A nell’appendice 2.
Valori mancanti
Il protocollo richiedeva la valutazione del caso osservato e l’ultima osservazione riportava i set di dati, quest’ultimo definitivo. L’ultima osservazione riportata sul metodo per correggere i valori mancanti era lo standard al momento in cui è stato condotto lo studio. Continua ad essere ampiamente utilizzato, sebbene i modelli più recenti come l’imputazione multipla o i modelli misti siano superiori. Abbiamo scelto di aderire al protocollo e di utilizzare il metodo dell’ultima osservazione riportata, inclusa l’imputazione multipla solo per confronto.
Variabili di risultato non specificate nel protocollo
C’erano quattro variabili di risultato nella CSR e nel documento pubblicato che non erano specificate nel protocollo. Queste erano le uniche misure di esito riportate come significative. Non sono stati inclusi in nessuna versione del protocollo come emendamenti (nonostante altri emendamenti), né sono stati presentati al comitato di revisione istituzionale. La CSR (sezione 3.9.1) afferma che facevano parte di un “piano di analisi” sviluppato circa due mesi prima che l’accecamento venisse interrotto. Nessun piano del genere appare nella CSR e non abbiamo documentazione contemporanea di tale affermazione, nonostante l’abbiamo ripetutamente richiesto a GSK.x
Conclusioni
Abbiamo deciso che la linea di azione migliore e più imparziale era analizzare i dati di efficacia nei dati dei singoli pazienti sulla base dell’ultima versione a priori garantita del protocollo SKB (1994, modificato nel 1996 per accettare una dimensione del campione ridotta). Sebbene il protocollo abbia omesso una discussione sulle correzioni che avremmo ritenuto necessarie, la correzione per più variabili è progettata per prevenire falsi positivi e non ci sono stati positivi. Eravamo d’accordo con i mandati statistici del protocollo, ma sebbene considerassimo inappropriati i confronti a coppie in assenza di significatività generale, riconosciamo che questa non è un’opinione universale, quindi abbiamo incluso i dati nella tabella A nell’appendice 2.
Infine, sebbene gli investigatori possano esplorare i dati come vogliono, variabili di esito aggiuntive al di fuori di quelle nel protocollo non possono essere legittimamente dichiarate una volta che lo studio è in corso, eccetto come “variabili esplorative”, appropriate per la discussione o come materiale per ulteriori studi ma non per l’analisi principale. Il protocollo a priori e l’accecamento sono le fondamenta di uno studio controllato randomizzato, garantendo che non vi sia nemmeno la possibilità del fenomeno HARK (“ipotesi dopo risultati noti”). Sebbene possiamo facilmente dimostrare che nessuna delle quattro variabili di esito non di protocollo dichiarate “positive” resiste all’esame accurato, il mandato principale dell’impresa RIAT è riaffermare le pratiche essenziali negli studi controllati randomizzati, quindi non abbiamo incluso queste variabili nel nostro analisi di efficacia.
Danni finali
Un’esperienza/evento avverso è stato definito nel protocollo (pagina 18) come “qualsiasi cambiamento nocivo, patologico o non intenzionale nelle funzioni anatomiche, fisiologiche o metaboliche come indicato da segni fisici, sintomi e/o cambiamenti di laboratorio che si verificano in qualsiasi fase della sperimentazione clinica se associato al farmaco o al placebo e se considerato correlato al farmaco o meno. Ciò include un’esacerbazione di condizioni o eventi preesistenti, malattie intercorrenti, interazione farmacologica o il peggioramento significativo della malattia in esame che non è registrato altrove nel modulo del caso clinico in base a valutazioni di efficacia specifiche “.
Gli eventi avversi dovevano essere ricavati dallo sperimentatore ponendo una domanda non determinante come: “Ti senti diverso in qualche modo dall’inizio del nuovo trattamento/dall’ultima valutazione?” Sono stati documentati i dettagli degli eventi avversi emersi con il trattamento, la loro gravità, inclusi eventuali cambiamenti nella somministrazione del farmaco in studio, l’attribuzione dello sperimentatore al farmaco in studio, qualsiasi terapia correttiva somministrata e lo stato dei risultati. L’attribuzione o la relazione con il farmaco in studio è stata giudicata dallo sperimentatore “non correlata”, “probabilmente non correlata”, “possibilmente correlata”, “probabilmente correlata” o “correlata”.
I segni vitali e gli elettrocardiogrammi sono stati ottenuti durante le visite settimanali. Ai pazienti con misure cardiovascolari potenzialmente preoccupanti è stata ridotta la dose del farmaco o sono stati ritirati dallo studio. Inoltre, se le concentrazioni sieriche combinate (ottenute alle settimane quattro e otto) di imipramina e desipramina superavano i 500 µg / ml, il paziente doveva essere ritirato dallo studio. Durante la visita di screening e alla fine dell’ottava settimana sono stati effettuati test clinici di laboratorio, tra cui chimica clinica, ematologia e analisi delle urine. Le anomalie di laboratorio clinicamente rilevanti dovevano essere incluse come eventi avversi.
Fonte dei dati sui danni
I dati sui danni in questo documento coprono la fase acuta, un periodo di scalaggio e una fase di follow-up fino a 30 giorni per coloro che hanno interrotto il trattamento a causa di eventi avversi. Per garantire la comparabilità con il report di Keller e colleghi, nessuna delle tabelle contiene dati della fase di prosecuzione.
I dati sugli eventi avversi provengono dalla CSR presentata sul sito web di GSK,19 principalmente nell’appendice D. L’appendice B fornisce dettagli sui farmaci concomitanti. Informazioni aggiuntive erano disponibili dalle descrizioni riassuntive nel corpo della CSR per i pazienti che hanno avuto eventi avversi che sono stati designati come gravi o hanno portato al ritiro. (Degli 11 pazienti che assumevano paroxetina che hanno manifestato eventi avversi designati come gravi, nove hanno interrotto il trattamento a causa di questi eventi). le violazioni, tuttavia, non hanno generato narrazioni di pazienti. Le tabelle nell’appendice D della CSR forniscono i termini letterali usati dagli investigatori in cieco, insieme ai termini preferiti come codificati da SKB utilizzando il dizionario ADECS (Avverse Drug Events Coding System). L’Appendice D include anche valutazioni di gravità e valutazioni di parentela. Abbiamo utilizzato il Medical Dictionary for Regulatory Activities (MedDRA) per codificare i termini letterali forniti nell’appendice D nella CSR.
La terminologia MedDRA è la terminologia medica internazionale sviluppata sotto gli auspici della Conferenza internazionale sull’armonizzazione dei requisiti tecnici per la registrazione dei prodotti farmaceutici per uso umano (ICH) www.meddra.org ), approvato dalla FDA e ora utilizzato da GSK. 20 Diverse limitazioni dei termini preferiti codificati ADECS forniti nell’appendice D della CSR sono diventati chiari quando abbiamo esaminato i termini preferiti ADECS assegnati ai termini letterali. In primo luogo, diversi termini letterali erano stati lasciati non codificati nell’ADECS. In secondo luogo, diversi eventi avversi riscontrati nelle narrazioni dei pazienti di eventi avversi gravi che hanno portato all’interruzione dello studio non sono stati trascritti nell’appendice D.
Abbiamo quindi contattato GSK per l’accesso ai moduli di segnalazione dei casi (appendice H della CSR), che non sono pubblicamente disponibili. GSK ha reso disponibili tutti i 275 moduli di case report per i pazienti inseriti nello studio 329. Questi moduli, tuttavia, per un totale di circa 77.000 pagine, erano disponibili solo tramite una struttura di desktop remoto (SAS Solutions OnDemand Secure Portal), 11 che lo rendeva difficile e richiede drasticamente molto tempo per ispezionare correttamente i record. 21 In effetti, solo una persona potrebbe intraprendere l’attività, con backup per casi ambigui. Di conseguenza non abbiamo potuto esaminare tutti i moduli di segnalazione dei casi. Invece abbiamo deciso di concentrarci su quegli 85 partecipanti identificati nelle appendici D e G della CSR che sono stati ritirati dallo studio, insieme ad altri otto partecipanti che erano noti dalla nostra ispezione delle CSR per essere diventati suicidi. Dei moduli di case report che sono stati controllati, 31 provenivano dal gruppo paroxetina, 40 dal gruppo imipramina e 22 dal gruppo placebo.
Tutti i moduli dei case report sono stati esaminati da JLN, che è stato addestrato all’uso di MedDRA. Il secondo revisore (JMN), un medico, non è stato formato nel sistema MedDRA, ma la formazione non è necessaria per la codifica dei dropout. Questi due revisori hanno concordato sui motivi per l’interruzione e la codifica degli effetti collaterali (non abbiamo utilizzato un indicatore quantitativo di accordo tra i valutatori). Abbiamo esaminato questi 93 moduli di case report per tutti gli eventi avversi che si sono verificati durante le fasi acuta, scalaggio e follow-up e abbiamo confrontato i nostri totali per gli eventi avversi con i totali riportati nell’appendice D della CSR. Questo processo di revisione ha identificato ulteriori eventi avversi che non erano stati registrati come termini letterali nell’appendice D della CSR. Ha anche portato alla ricodifica di molti dei motivi per l’interruzione. Tabelle B, C, Mancavano almeno 1000 pagine dai moduli di segnalazione del caso che abbiamo esaminato, senza alcun motivo distinguibile per le informazioni mancanti: ad esempio, un modulo veniva fornito con una pagina inserita in cui si dichiarava che le pagine da 114 a 223 mancavano, senza indicare i motivi.
Codifica degli eventi avversi
Scelta del dizionario di codifica per i danni
Il protocollo (pagina 25) indica che gli eventi avversi dovevano essere codificati e confrontati in base al termine preferito e al sistema corporeo utilizzando statistiche descrittive, ma non prespecifica una scelta del dizionario di codifica per generare termini preferiti da termini letterali. La CSR (scritta al termine dello studio) specifica che gli eventi avversi rilevati dagli investigatori clinici in questo studio sono stati codificati con ADECS, che all’epoca era utilizzato da SKB. Questo sistema è stato derivato da un sistema di codifica sviluppato dalla Food and Drug Administration (FDA) statunitense, Coding Symbols for a Thesaurus of Adverse Reaction Terms (COSTART), ma l’ADECS non è di per sé un sistema riconosciuto e non è più disponibile.
Abbiamo codificato gli eventi avversi utilizzando MedDRA, che ha sostituito COSTART per la FDA perché è di gran lunga il sistema di codifica più comunemente usato oggi. Ai fini della codifica, abbiamo preso i termini originali usati dagli investigatori clinici, come trascritti nell’appendice D della CSR, e abbiamo applicato i codici MedDRA a queste descrizioni. Le informazioni dell’appendice D sono state trascritte in fogli di calcolo (disponibili su www.Study329.org). I termini letterali ei termini di codifica ADECS sono stati trascritti per primi in questi fogli, consentendo a tutta la codifica di essere eseguita prima che i nomi dei farmaci fossero aggiunti. La trascrizione è stata eseguita da un assistente di ricerca che era un codificatore addestrato da MedDRA ma non ha preso parte al codifica effettiva.
Tutta la codifica è stata eseguita da JLN e controllata da DH o viceversa. Tutta la nostra codifica dai termini letterali nell’appendice D della CSR è stata eseguita in cieco, così come la codifica dai moduli di segnalazione dei casi. Presentiamo i risultati come SKB li ha presentati nella CSR utilizzando il dizionario ADECS (tabella 14.2.1) e come codificati da noi utilizzando MedDRA. In generale, la codifica MedDRA rimane più vicina dell’ADECS alla descrizione originale dell’evento da parte del medico. Ad esempio, MedDRA codifica “mal di gola” come “mal di gola” ma SKB, utilizzando ADECS, lo ha codificato come “faringite” (infiammazione della gola). Il mal di gola può insorgere a causa della faringite, ma quando qualcuno sta assumendo inibitori selettivi della ricaptazione della serotonina possono indicare una reazione distonica nell’area orofaringea 22
La classificazione di un problema come “disturbo del sistema respiratorio” (infiammazione) piuttosto che come “distonia” (un disturbo del sistema nervoso centrale) può fare una differenza considerevole per il profilo degli eventi avversi apparenti di un farmaco. Rimanendo più vicino alla descrizione originale degli eventi, MedDRA codifica gli eventi suicidi come “ideazione suicidaria” o “autolesionismo / tentato suicidio” piuttosto che l’opzione ADECS di “labilità emotiva”; allo stesso modo, l’aggressività è più chiaramente contrassegnata come “eventi aggressivi” piuttosto che “ostilità”.
La maggior parte del codice era semplice. Quasi tutti i termini letterali semplicemente mappati sui termini di codifica in MedDRA. Le sfide di codifica di solito erano correlate a casi in cui si erano verificati eventi avversi significativi, ma i pazienti erano stati designati da SKB per aver interrotto il trattamento per mancanza di efficacia. Non c’era alcuna narrativa del paziente per tali pazienti, a differenza dei pazienti che si ritiene abbiano interrotto il trattamento a causa dell’evento avverso che si è verificato alla sospensione. C’erano poche decisioni di programmazione impegnative. L’appendice 3 mostra la nostra codifica dei casi in cui sono stati considerati comportamenti suicidari e autolesionistici.
Analisi dei dati sui danni
Nell’analizzare i dati sui danni per la popolazione responsabile della sicurezza, abbiamo innanzitutto esplorato le discrepanze nel numero di eventi tra i moduli di segnalazione dei casi e la CSR. In secondo luogo, abbiamo presentato tutti gli eventi avversi piuttosto che quelli che si verificano solo a una velocità particolare (come fatto da Keller e colleghi). In terzo luogo, abbiamo raggruppato gli eventi in gruppi più ampi di classificazione per sistemi e organi (SOC): psichiatrici, cardiovascolari, gastrointestinali, respiratori e altri. La Tabella D nell’appendice 2 riassume tutti gli eventi avversi per tutti i gruppi SOC MedDRA. In quarto luogo, abbiamo suddiviso gli eventi in base alla gravità, selezionando gli eventi avversi codificati come gravi e utilizzando l’elenco nell’appendice G della CSR dei pazienti che hanno interrotto il trattamento per qualsiasi motivo. In quinto luogo, abbiamo incluso un’analisi degli effetti del trattamento precedente, presentando i profili della fase di run-in dei farmaci assunti dai pazienti che entrano in ciascuno dei tre bracci dello studio e confrontando l’elenco degli eventi avversi sperimentati dai pazienti trattati con farmaci concomitanti (dall’appendice B) rispetto a quelli non trattati con altri farmaci. Infine, abbiamo estratto gli eventi che si sono verificati durante la fase di scalaggio e follow-up. Non abbiamo intrapreso test statistici sui dati sui danni, come discusso di seguito.
Ritiro del paziente
Un paziente in studio potrebbe ritirarsi o essere ritirato prematuramente per “esperienze avverse inclusa malattia intercorrente”, “effetto terapeutico insufficiente”, “deviazione dal protocollo inclusa la non conformità”, “perdita al follow-up”, “interruzione da parte di SB [SKB], “E” altro (specificare)”. La CSR afferma che il motivo principale del ritiro è stato determinato dallo sperimentatore. Abbiamo esaminato i codici forniti per l’interruzione dello studio, che si trovano nell’appendice G della CSR, e abbiamo apportato modifiche in una parte dei casi.
Metodi statistici
La popolazione di interesse primaria era l’intenzione di trattare la popolazione che includeva tutti i pazienti che avevano ricevuto almeno una dose del farmaco in studio e avevano almeno una valutazione di efficacia dopo il basale. Le caratteristiche demografiche, la descrizione dell’episodio depressivo di base, ulteriori diagnosi psichiatriche e le variabili della storia personale dei pazienti sono state riassunte descrittivamente per gruppo di trattamento.
L’endpoint della fase acuta otto settimane era il nostro interesse principale. Le conclusioni statistiche sull’efficacia di paroxetina e imipramina sono state tratte utilizzando i dati ottenuti dall’ultima osservazione portata avanti (cioè l’ultima valutazione “in terapia” durante la fase acuta) e da set di dati di casi osservati. Paroxetina e imipramina dovevano essere confrontate ciascuna con il placebo; non doveva esserci confronto tra paroxetina e imipramina.
Abbiamo seguito i metodi del protocollo di studio a priori del 1994 (modificato nel 1996 per accettare una dimensione del campione ridotta). Non ha fornito ipotesi statistiche esplicite (ipotesi nulle e ipotesi alternative); né c’erano giustificazioni per gli approcci statistici proposti o le ipotesi statistiche sottostanti.
Una delle due variabili di efficacia primarie, la proporzione di responder (risposta) e una variabile di efficacia secondaria, la percentuale di pazienti recidivanti, sono state trattate come variabili categoriali. La seconda variabile primaria di efficacia, la variazione del punteggio totale HAM-D durante la fase acuta e le rimanenti variabili secondarie di efficacia sono state trattate come variabili continue.
In accordo con il protocollo, le variabili continue sono state analizzate con l’analisi parametrica della varianza (ANOVA) con effetti nel modello inclusi trattamento, sperimentatore e trattamento per interazione sperimentatore. I confronti a coppie non sono stati eseguiti se l’ANOVA omnibus (complessiva) non era significativa (P bilaterale <0,05), come specificato dal protocollo (riconosciamo opinioni divergenti su questo problema nella letteratura statistica, 23 quindi li abbiamo inclusi nella tabella A in appendice 2, per completezza). Le variabili categoriali sono state analizzate con regressione logistica, includendo gli stessi effetti. In entrambi i casi, se il trattamento per interazione dello sperimentatore ha portato a una P bilaterale> 0,10, il termine di interazione è stato eliminato dal modello.
I test statistici sono stati eseguiti con le procedure del modello lineare (LM) e dei modelli lineari generali (GLM) del pacchetto statistico R (versione 2.15.2) come fornito da GSK. L’assegnazione è stata eseguita con il pacchetto di imputazione multipla mediante equazioni concatenate (MICE) anche in R. 24 Per le analisi dei tassi di ricaduta, sono stati inclusi tutti i responder (HAM-D ≤8 o riduzione ≥50% dei sintomi) che soddisfacevano i criteri originali per l’accesso alla fase di continuazione dello studio. I pazienti erano considerati recidivanti se non soddisfacevano più i criteri di risposta o se erano stati ritirati per “sovradosaggio intenzionale”.
Risultati
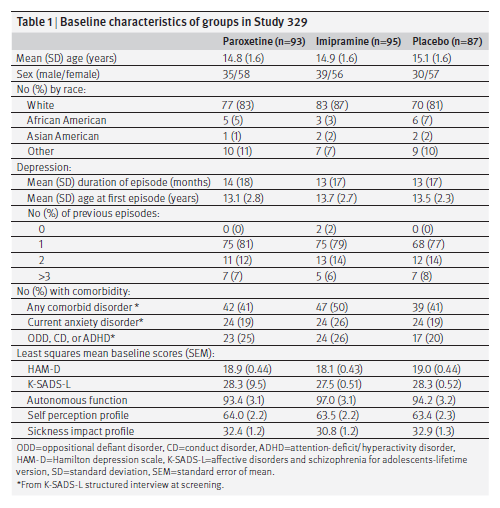
La tabella 1 mostra i dati demografici dei gruppi, insieme ai parametri di depressione, comorbidità e punteggi di base per le variabili di efficacia.
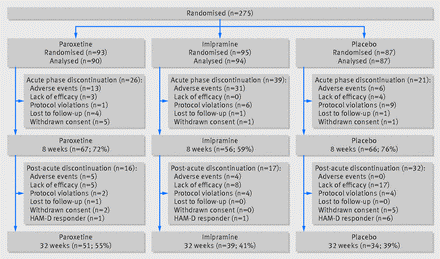
La Figura 1 riassume le allocazioni e le interruzioni tra i tre gruppi di trattamento durante il periodo di studio acuto. Il diagramma di flusso copre l’intenzione di trattare la popolazione per la fase acuta e l’analisi di efficacia. Il gruppo paroxetina è stato titolato a una dose di 20 mg/giorno entro la quarta settimana, con il 55% (51/93) dei partecipanti che è passato a una dose più alta (media 28,0 mg/giorno, SD 8,4 mg) entro la settimana otto. Il gruppo imipramina è stato titolato a 200 mg / die entro la quarta settimana, con il 40% (38/95) che è passato a una dose più alta (media 205,8 mg / die, DS 63,9 mg) entro l’ottava settimana. Ventotto pazienti hanno raggiunto la dose massima consentita di 40 mg di paroxetina e 20 pazienti sono stati titolati fino a un massimo di 300 mg di imipramina.
Efficacia
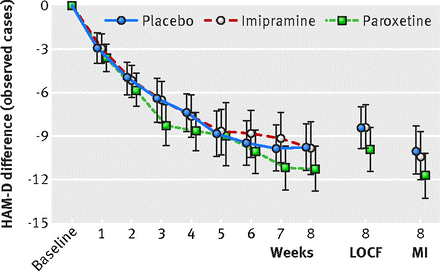
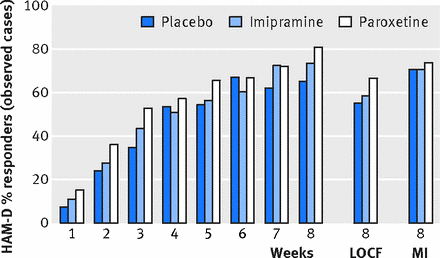
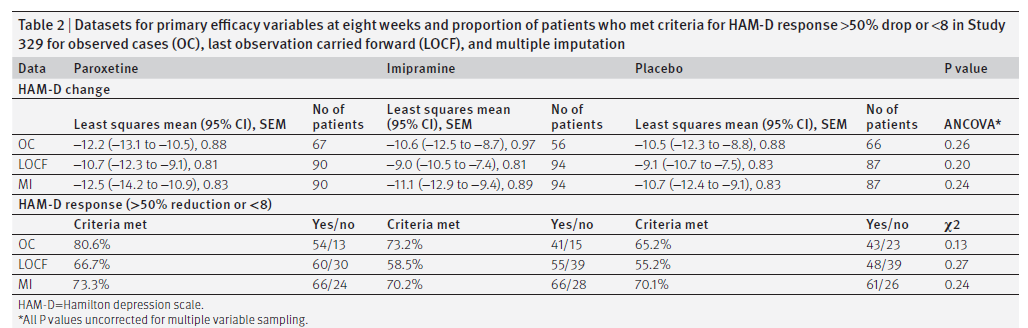
Non ci sono state discrepanze tra nessuna delle nostre analisi e quelle contenute nella CSR. Le figure 2 e 3 illustrano i valori longitudinali per le due variabili di efficacia primarie: variazione media dal basale nel punteggio HAM-D e percentuale di risposta, definita come una diminuzione del punteggio HAM-D del 50% o più rispetto al basale o punteggio finale HAM-D di ≤8. La differenza tra paroxetina e placebo è scesa al di sotto del livello di significatività clinica prespecificato (4 punti) e nessuno dei due risultati primari ha raggiunto significatività a qualsiasi intervallo misurato per qualsiasi set di dati durante la fase acuta. La rianalisi formale includeva sia i casi osservati che i set di dati riportati dell’ultima osservazione. Come accennato in precedenza, il set di dati di assegnazione multipla è incluso per il confronto. Non c’era significatività statistica (considerata a P <0,05) o significatività clinica mostrata per nessuna delle variabili di efficacia primarie o secondarie prespecificate nel caso osservato o nell’ultima osservazione riportata nei set di dati, quindi l’analisi a coppie è stata considerata ingiustificata. La Tabella 2 mostra i risultati alla settimana otto per la riduzione del punteggio HAM-D e per la percentuale di pazienti che hanno soddisfatto i criteri per la risposta HAM-D. I punteggi HAM-D sono diminuiti di 10,7 (intervallo di confidenza al 95% da 9,1 a 12,3), 9,0 (da 7,4 a 10,5) e 9,1 (da 7,5 a 10,7) punti (media dei minimi quadrati) per i gruppi paroxetina, imipramina e placebo, rispettivamente.
Tavolo 2
Set di dati per le variabili di efficacia primaria a otto settimane e percentuale di pazienti che hanno soddisfatto i criteri per la risposta HAM-D calo> 50% o <8 nello studio 329 per i casi osservati (OC), ultima osservazione portata a termine (LOCF) e imputazione multipla.
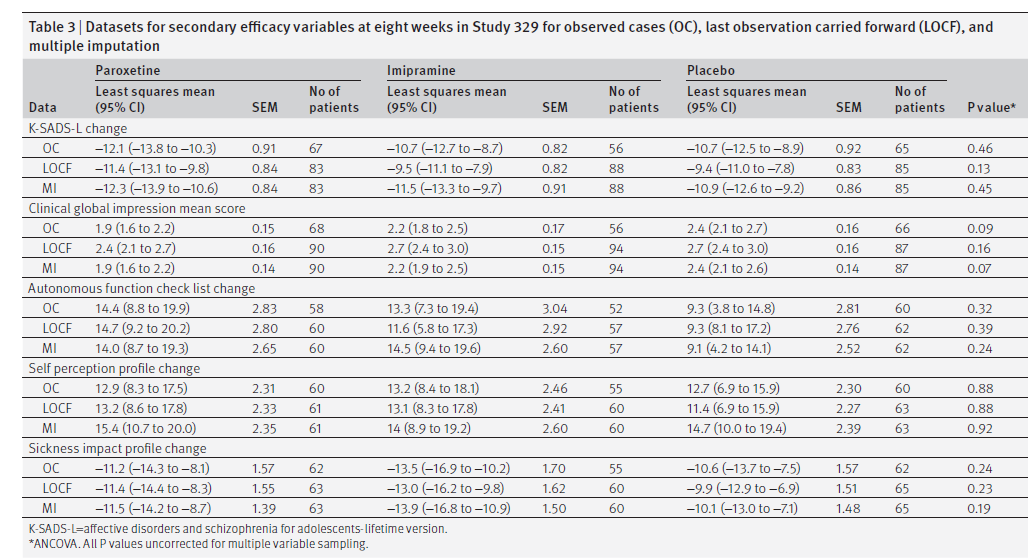
La tabella 3 mostra i risultati a otto settimane per le variabili di efficacia secondarie.
Tabella 3
Sebbene il protocollo elencasse i “predittori di risposta” tra le variabili di efficacia secondarie, l’assenza di differenze statisticamente o clinicamente significative tra i tre bracci ha reso nulla questa analisi. Il protocollo ha anche elencato il tasso di ricaduta nella fase di continuazione per i responder come variabile di esito secondaria. Il nostro calcolo differiva da quello del CSR perché includevamo quelli i cui punteggi HAM-D erano superiori al range di “risposta” e quelli che avevano intenzionalmente overdose. Nella fase di continuazione, i tassi di abbandono erano troppo alti in tutti i gruppi per una interpretazione precisa: 33/51 (65%) nel gruppo paroxetina; 25/39 (64%) nel gruppo imipramina; e 21/34 (62%) nel gruppo placebo. Le ricadute registrate sono state rispettivamente 25/51 (49%), 16/39 (41%) e 12/34 (35%). Sebbene il tasso di ricaduta fosse inferiore nel gruppo placebo, le differenze non erano significative (2 × 3 χ 2 P = 0,44). Set di dati per variabili di efficacia secondarie a otto settimane nello studio 329 per casi osservati (OC), ultima osservazione portata avanti (LOCF) e imputazione multipla.
Danni
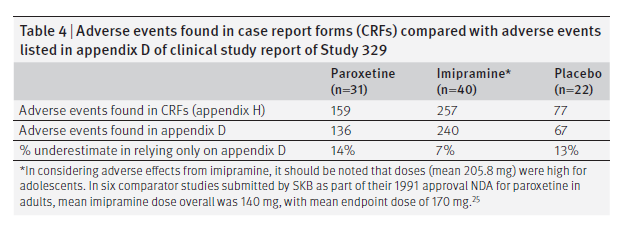
Revisione dei moduli di segnalazione dei casi
Abbiamo esaminato i moduli di case report nell’appendice H per 93 (34%) di 275 pazienti. Abbiamo scoperto eventi avversi registrati sui moduli di segnalazione del caso ma non trascritti negli elenchi di eventi avversi a livello di paziente nell’appendice D della CSR. La tabella 4 mostra queste discrepanze. Le categorie più comuni di eventi avversi aggiuntivi riscontrati nei moduli di segnalazione dei casi erano psichiatrici per paroxetina (12/23) e placebo (4/10) e cardiovascolari per imipramina (5/17) (tabella B nell’appendice 2).
Codifica e rappresentazione dei dati sugli eventi avversi
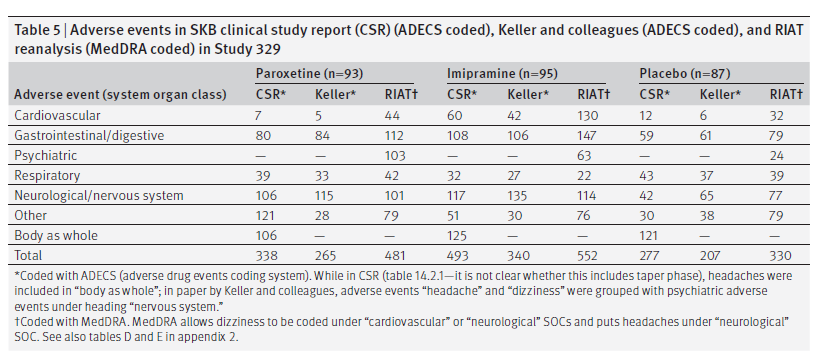
La Tabella 5 presenta il numero di eventi avversi riscontrati in questo studio riassunti per classificazione per sistemi e organi (SOC), in primo luogo come codificato da SKB utilizzando ADECS, in secondo luogo come riportato da Keller e colleghi (che hanno segnalato solo eventi avversi che si sono verificati con una frequenza superiore al 5%) e in terzo luogo come codificato da noi utilizzando MedDRA. Alcuni eventi avversi rientrano sempre in una particolare classe per sistemi e organi; altri richiedono che il programmatore scelga tra classi per sistemi e organi. Un elenco completo degli eventi avversi può essere trovato nella tabella E nell’appendice 2.
Abbiamo incluso gli eventi che si verificano durante la fase di scalaggio che SKB ha assegnato alla fase di continuazione come eventi avversi di fase acuta. In uno studio che ha una fase di prosecuzione, la valutazione degli eventi avversi solleva una difficoltà metodologica non ancora affrontata da gruppi come CONSORT. Se uno studio ha solo una fase acuta, vengono conteggiati tutti gli eventi avversi per tutti i pazienti che ricevono il trattamento così come in qualsiasi fase di scalaggio e spesso per un periodo di follow-up di 30 giorni. Quando uno studio ha una fase di continuazione, i periodi di scalaggio e di follow-up di 30 giorni vengono spostati. Per garantire un’analisi comparabile di tutti i partecipanti, abbiamo conteggiato gli eventi avversi nella fase acuta e in entrambe le fasi di scalaggio e follow-up, spiazzati o meno. SKB non sembra averlo fatto, portando ad alcune differenze nei numeri.
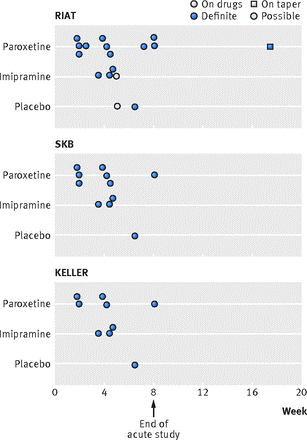
Fig 4 Tempistica degli eventi suicidari e autolesionistici nello studio 329, Keller e colleghi e nell’analisi RIAT
La tabella 6 mostra il numero di comportamenti suicidari e autolesionistici che abbiamo identificato nella nostra analisi RIAT e confrontati con quanto riportato da Keller e colleghi e documentato nella CSR (tabella 6). I dettagli completi per i pazienti inclusi in questa tabella possono essere trovati nell’appendice 3, insieme alle note di lavoro e alle indicazioni su dove si possono trovare i dettagli chiave nella CSR. È possibile adottare approcci diversi per spostare gli eventi della fase di scalaggio nella fase di continuazione e rivedere la codifica per tutti i casi, in particolare i casi 039, 089 e 106, che sono stati designati comportamenti suicidari e autolesionistici nella ricodifica RIAT. Ciò comporterebbe cifre diverse. Non ci sono stati cambiamenti degni di nota nei dati fisiologici, che sono dettagliati nell’appendice F (elenchi dei dati dei pazienti dei test di laboratorio) nella CSR.
| table 6 | numbers of patients with suicidal and self injurious behaviours in study 329 with different safety methods |
| Paroxetine (n=93) imipramine (n=95) Placebo (n=87) |
| Keller and colleagues* 5 3 1 |
| SKB acute from CSR* 7 3 1 |
| RIAT acute and taper from CSR 11 4 (3 definite, 1 possible) 2 (1 definite, 1 possible) |
| *Keller and colleagues and CSR mostly reported suicide related events as “emotional lability.” |
Livelli di gravità
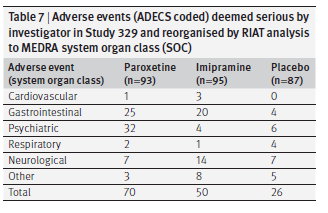
Nella CSR, eventi avversi gravi (definiti come un evento che “ha provocato il ricovero in ospedale, è stato associato a gesti suicidi o è stato descritto come grave dal medico curante”) sono stati riportati in 11 pazienti nel gruppo paroxetina, cinque nel gruppo imipramina e due nel gruppo placebo. Designare un evento avverso come grave dipendeva dal giudizio dello sperimentatore clinico. Pertanto non siamo stati in grado di esprimere giudizi comparabili sulla gravità, ma esistono altri due metodi per affrontare la questione della gravità degli eventi avversi. Uno è quello di considerare quelli classificati come gravi piuttosto che moderati o lievi al momento dell’evento (tabella 7). Un numero e una proporzione elevati di eventi psichiatrici gravi si sono verificati nel gruppo paroxetina. Al contrario, pochi dei molti eventi cardiovascolari nel gruppo imipramina sono stati classificati come gravi.
Interruzioni
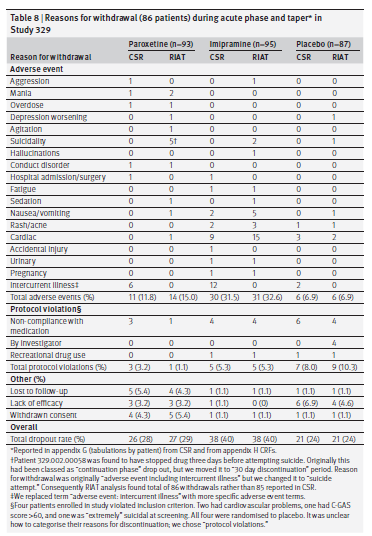
Un secondo metodo per affrontare la questione della gravità degli eventi avversi è esaminare i tassi di interruzione a causa di tali eventi. La Tabella 8 mostra i motivi del ritiro durante la fase acuta e di scalaggio graduale a causa di eventi avversi e altre cause. Si noti che abbiamo esaminato i moduli dei rapporti sui casi dell’appendice H per tutte le interruzioni riportate nell’appendice G della CSR. Tutte le modifiche alla codifica per l’interruzione sono riportate nella tabella H nell’appendice 2. Considerazione sullo scalaggio rimosso nello studio 329 ha rivelato un enigma. Oltre agli 86 abbandoni dalla fase acuta rilevati da SKB, ci sono stati 65 abbandoni dopo che le valutazioni sono state completate alla settimana otto. SKB considerava questi pazienti come partecipanti alla fase di continuazione, sebbene nessuno di loro avesse assunto un farmaco in fase di continuazione o avesse una valutazione della fase di continuazione. La codifica per l’interruzione era particolarmente ambigua per questo gruppo.
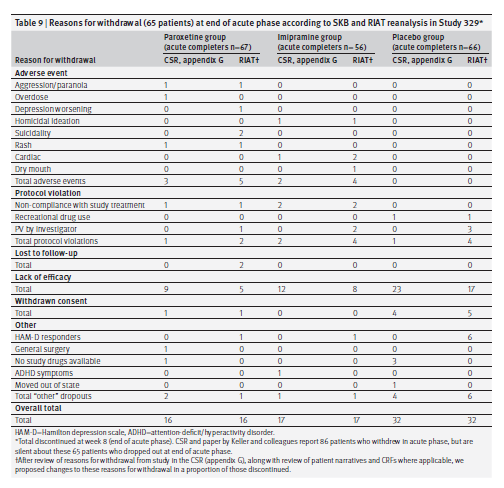
La maggior parte dei pazienti interrotti a questo punto sono stati designati da SKB come “mancanza di efficacia” (tabella 9). I ricercatori in quattro centri hanno segnalato la mancanza di efficacia come motivo per interrompere sei pazienti assegnati al placebo anche se il punteggio HAM-D era nel range dei responder e in alcuni casi era di 2 o 3 punti. In alcuni casi si sono verificate evidenti violazioni del protocollo o fattori come l’indisponibilità di ulteriori trattamenti (placebo in particolare). Abbiamo ricategorizzato la mancanza di abbandoni di efficacia in base a fattori come eventi avversi e punteggi HAM-D. La tabella 9 mostra la nostra analisi dei motivi del ritiro alla fine della fase acuta.
Effetti di sospensione
Il protocollo per lo studio 329 ha richiesto una fase di scalaggio per tutti i partecipanti e, inoltre, un periodo di follow-up di 30 giorni per tutti coloro che hanno interrotto lo studio a causa di eventi avversi. I dati nell’appendice D della CSR consentono di identificare gli eventi avversi che si verificano nei periodi di scalaggio e follow-up. Questi dati sono presentati nella tabella 10.
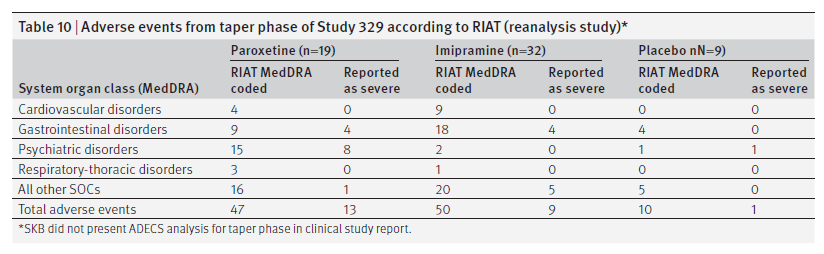
Effetti di altri farmaci
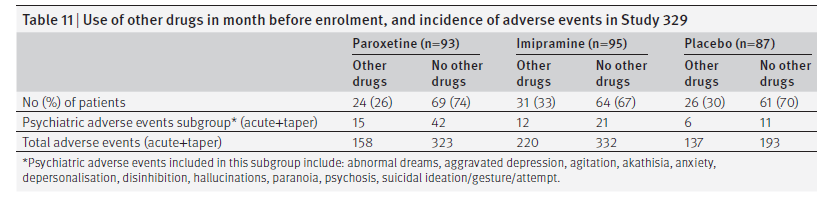
La tabella 11mostra i dati sugli effetti di altri farmaci sugli eventi avversi registrati. I pazienti che assumevano altri farmaci hanno avuto più eventi avversi rispetto a quelli che non lo erano. Questo effetto è stato leggermente più marcato nel gruppo placebo, e come tale funziona con l’apparente beneficio dei trattamenti farmacologici attivi nel ridurre al minimo qualsiasi eccesso di eventi avversi rispetto al placebo.
Discussione
Principali risultati e confronto con la pubblicazione originale della rivista
La nostra analisi RIAT dello studio 329 ha mostrato che né la paroxetina né l’imipramina ad alte dosi erano efficaci nel trattamento della depressione maggiore negli adolescenti e c’era un aumento clinicamente significativo dei danni con entrambi i farmaci. Questa analisi contrasta sia con le conclusioni pubblicate di Keller e colleghi 2 sia con il modo in cui i risultati sono stati riportati e interpretati nella CSR. Abbiamo analizzato e riportato lo studio 329 secondo il protocollo originale (con emendamenti approvati). L’appendice 1 mostra le fonti di informazione che abbiamo usato nella preparazione di questo documento, che dovrebbe aiutare altri ricercatori che vogliono accedere ai dati per controllare la nostra analisi o per interrogarli in altri modi. Traiamo conclusioni minime sull’efficacia e sui danni, invitando gli altri a offrire la propria analisi.
Il nostro riesame dei dati, inclusa una revisione del 34% dei casi, non ha mostrato discrepanze significative nei dati di efficacia primaria. La marcata differenza tra gli esiti di efficacia riportati da noi e quelli riportati da SKB deriva dal fatto che la nostra analisi si è mantenuta fedele ai metodi del protocollo e alla sua designazione di variabili di esito primarie e secondarie.
Gli autori/sponsor si sono discostati dal loro protocollo di studio nella stessa CSR eseguendo confronti a coppie di due dei tre gruppi quando l’ANOVA omnibus non ha mostrato alcun significato nelle variabili continue o dicotomiche. Hanno anche segnalato come significative altre quattro variabili che non erano state menzionate nel protocollo o nei suoi emendamenti, senza alcun riconoscimento che queste misure fossero state introdotte post hoc. Questa disposizione contravveniva alla disposizione II dell’appendice B del protocollo dello Studio 329 (“Questioni amministrative”), secondo la quale qualsiasi cambiamento al protocollo dello studio doveva essere presentato come emendamento/modifica.
Per quanto riguarda gli eventi avversi, c’erano differenze ampie e clinicamente significative tra i dati da noi analizzati, quelli riassunti nella CSR utilizzando i metodi ADECS e quelli riportati da Keller e colleghi. Queste differenze derivano dall’inserimento inadeguato e incompleto dei dati dai moduli di segnalazione dei casi alle schede di dati di riepilogo nella CSR, dal sistema di codifica ADECS utilizzato da SKB e dalla segnalazione di queste schede dati in Keller e colleghi. SKB ha riportato 338 eventi avversi con paroxetina e Keller e colleghi ne hanno segnalati 265, mentre ne abbiamo identificati 481 dalla nostra analisi della CSR e abbiamo trovato altri 23 che erano stati persi dai 93 moduli di case report che abbiamo esaminato.
Un altro motivo per cui le cifre di Keller e colleghi sono inferiori alle nostre è perché hanno presentato dati solo per eventi avversi segnalati per il 5% o più dei pazienti. Per tutti gli eventi avversi combinati, la loro tabella 3 riportava un carico di eventi avversi con paroxetina 1,2 volte quello del carico con placebo. Questo confronta con la figura di 1.4 dalla nostra codifica RIAT MedDRA dei dati dalla CSR. Anche le cifre dei moduli CSR e case report differiscono sostanzialmente da altre cifre citate da Keller e colleghi perché non hanno segnalato una categoria di eventi avversi psichiatrici, ma hanno invece raggruppato tali eventi insieme a “vertigini” e “mal di testa” sotto la classe sistema nervoso.”
MedDRA distingue tra classi per sistemi e organi neurologiche e psichiatriche. Abbiamo inserito il mal di testa nella classe neurologica piuttosto che in quella psichiatrica. MedDRA consente di codificare le vertigini in classi cardiovascolari o neurologiche. Data la dose di imipramina utilizzata, la maggior parte dei casi di vertigini sembra essere cardiovascolare, con Keller e colleghi che riportano anche un alto tasso di ipotensione posturale con imipramina. Abbiamo quindi codificato tutte le vertigini in ambito cardiovascolare piuttosto che neurologico. C’è spazio per altri che accedono ai dati per analizzare se ci sono informazioni sufficienti per codificare certi casi di vertigini, come vertigini durante lo scalaggio della paroxetina, come neurologici, ma non abbiamo effettuato quell’analisi più complessa.
Come riportato da Keller e colleghi, vertigini e mal di testa comprendevano 54 su 115 eventi del sistema nervoso in quelli che assumevano paroxetina (47%), 83 su 135 eventi in quelli che assumevano imipramina (62%) e 50 su 65 eventi in quelli che assumevano placebo (77 %). L’effetto di districare questi due sintomi dagli eventi avversi psichiatrici smaschera una differenza clinicamente importante nei profili degli eventi avversi psichiatrici tra paroxetina e placebo.
C’era una grande differenza tra la frequenza del pensiero suicida e degli eventi riportati da Keller e colleghi e la frequenza documentata nella CSR, come mostrato nella tabella 6. Per quanto riguarda gli abbandoni, Keller e colleghi hanno affermato che il 69% dei pazienti ha completato la fase acuta. Solo il 45%, invece, è passato alla fase di prosecuzione, che non è stata ancora oggetto di analisi RIAT.
Confronto con altri studi
I nostri risultati sono coerenti con quelli di altri studi, incluso un recente esame di 142 studi su sei farmaci psicotropi per i quali erano disponibili sia articoli di riviste che sommari di studi clinici. 26,27 La maggior parte dei decessi (94/151, 62%) e dei suicidi (8/15, 53%) riportati nelle sintesi degli studi non sono stati riportati negli articoli di giornale. Solo uno dei nove suicidi negli studi sull’olanzapina è stato segnalato in articoli pubblicati.
Segnalazione di eventi avversi
La nostra nuova analisi dello Studio 329 ha mostrato notevoli variazioni nel modo in cui possono essere segnalati gli eventi avversi, dimostrando diversi modi in cui l’analisi e la presentazione dei dati di sicurezza possono influenzare l’apparente sicurezza di un farmaco. Abbiamo identificato i seguenti potenziali ostacoli a una segnalazione accurata dei danni (riassunti nel riquadro 2):
Riquadro 2: potenziali ostacoli a una segnalazione accurata dei danni
- Uso di un sistema di codifica idiosincratico
- Mancata trascrizione di tutti gli eventi avversi dalla cartella clinica al database degli eventi avversi
- Filtraggio dei dati sugli eventi avversi attraverso tecniche statistiche
- Limitazione della segnalazione agli eventi che si sono verificati al di sopra di una data frequenza in un qualsiasi gruppo
- Evento di codifica sotto diversi titoli per diversi pazienti (diluizione)
- Raggruppamento di eventi avversi
- Considerazione insufficiente della gravità
- Codifica della correlazione al farmaco in studio
- Effetti mascheranti di farmaci concomitanti
- Ignorare gli effetti della sospensione del farmaco
Uso di un sistema di codifica idiosincratico
Il termine “labilità emotiva”, come utilizzato nel sistema di codifica degli eventi avversi da farmaco della SKB, maschera le differenze nel comportamento suicidario tra paroxetina e placebo.
Mancata trascrizione di tutti gli eventi avversi dalla cartella clinica al database degli eventi avversi
La nostra revisione dei moduli di case report ha rivelato una significativa sotto-registrazione di eventi avversi.
Filtraggio dei dati sugli eventi avversi attraverso tecniche statistiche
Keller e colleghi (e GSK nella successiva corrispondenza) hanno ignorato i dati sui danni sfavorevoli sulla base del fatto che la differenza tra paroxetina e placebo non era statisticamente significativa, in contrasto con il protocollo SKB che richiedeva confronti primari da effettuare utilizzando statistiche descrittive. A nostro avviso, statisticamente significativi o meno, tutti gli esiti primari e secondari rilevanti e gli esiti dannosi dovrebbero essere riportati esplicitamente. Il test per la significatività statistica è più appropriato intrapreso per le misure di esito primarie poiché il potere dello studio si basa su queste. Non abbiamo effettuato test statistici per i danni poiché non conosciamo alcun modo valido per interpretarli. Per allontanarsi da una presentazione dicotomica (significativa/non significativa) di prove, abbiamo deciso di presentare tutte le prove originali e ricodificate per consentire ai lettori la propria interpretazione. I dati presentati in appendice 2 e relative schede di lavoro depositate su www.Study329.org consentirà prontamente altri approcci all’analisi dei dati per coloro che sono interessati, e accogliamo con favore altre analisi.
Limitazione della segnalazione agli eventi che si sono verificati al di sopra di una data frequenza in un qualsiasi gruppo
Nel documento di Keller e colleghi, riportare solo gli eventi avversi che si sono verificati in più del 5% dei pazienti ha oscurato l’onere dei danni. Al contrario, segnaliamo tutti gli eventi avversi che sono stati registrati. Questi sono disponibili nella tabella E nell’appendice 2.
Evento di codifica sotto diversi titoli per diversi pazienti (diluizione)
L’effetto di segnalare solo eventi avversi che hanno una frequenza superiore al 5% è aggravato quando, per esempio, l’agitazione può essere codificata sotto agitazione, ansia, nervosismo, ipercinesia e labilità emotiva; quindi, un problema che si verifica a un tasso > 10% potrebbe svanire essendo codificato in sottotitoli diversi in modo tale che nessuno di questi raggiunga un tasso di soglia del 5%.
Oltre a rendere disponibili tutti i dati in modo che altri possano esaminarli, un modo per compensare questa possibilità è presentare tutti i dati in gruppi più ampi di classi di sistemi e organi. MedDRA offre i seguenti livelli superiori: psichiatrico, cardiovascolare, gastrointestinale, respiratorio e altro. Nella tabella E nell’appendice 2, gli eventi avversi codificati qui sotto “altro” sono suddivisi sotto le voci aggiuntive del SOC MedDRA, inclusi generale, sistema nervoso, metabolico e gravidanza.
Raggruppamento di eventi avversi
Anche quando sono presentati in gruppi di sistemi più ampi, raggruppare sintomi comuni e benigni con quelli più importanti può mascherare problemi di sicurezza. Ad esempio, nel documento di Keller e colleghi, eventi avversi comuni come vertigini e mal di testa sono raggruppati con eventi avversi psichiatrici nell’intestazione SOC “sistema nervoso”. Poiché questi eventi avversi sono comuni tra i bracci di trattamento, questo raggruppamento ha l’effetto di diluire la differenza negli effetti collaterali psichiatrici tra paroxetina, imipramina e placebo.
Abbiamo seguito MedDRA nel segnalare vertigini in eventi “cardiovascolari” e mal di testa in “sistema nervoso”. Potrebbero esserci categorie migliori; il nostro raggruppamento è provvisorio piuttosto che strategico. Nella tabella E nell’appendice 2, abbiamo elencato tutti gli eventi codificati sotto ogni intestazione di classe per sistemi e organi e invitiamo altri a esplorare ulteriormente questi problemi, inclusa la categorizzazione alternativa di livello superiore di questi eventi avversi.
Considerazione insufficiente della gravità
Oltre a codificare gli eventi avversi, i ricercatori li classificano in base alla gravità. Se non viene fatto alcun tentativo di tenere conto della gravità e di includerla nella segnalazione, i lettori potrebbero avere l’impressione che ci fosse un uguale carico di eventi avversi in ciascun braccio, quando in realtà tutti gli eventi in un braccio potrebbero essere gravi e duraturi mentre quelli nell’altro potrebbe essere lieve e transitorio. Un modo per gestirlo è guardare in modo specifico a quei pazienti che abbandonano lo studio a causa di eventi avversi. Un altro metodo consiste nel segnalare quegli eventi avversi codificati come gravi per ciascun gruppo di farmaci separatamente da quelli codificati come lievi o moderati. Abbiamo utilizzato entrambi gli approcci (vedi tabelle 7 e 8).
Codifica della correlazione al farmaco in studio
I giudizi degli investigatori sul fatto che un evento avverso sia correlato al farmaco possono portare a scartare l’importanza di un effetto. Abbiamo incluso questi giudizi nei fogli di lavoro depositati su www.Study329.org, ma non li abbiamo analizzati perché è diventato chiaro che l’accecamento era stato rotto in diversi casi prima che la connessione fosse giudicata dagli investigatori originali e perché alcuni giudizi non erano plausibili. Ad esempio, è documentato a pagina 279 nella CSR che un ricercatore, sapendo che il paziente era sotto placebo, ha dichiarato che un evento suicida era “decisamente correlato al trattamento” sulla base del fatto che “il peggioramento della depressione e il pensiero suicida erano in pericolo di vita e sicuramente correlato al farmaco in studio [noto per essere placebo] in quanto c’era una mancanza di effetto”. In particolare, degli 11 pazienti con eventi avversi gravi con paroxetina (rispetto a due con placebo) riportati nel documento da Keller e colleghi, solo uno “è stato considerato dallo sperimentatore curante correlato al trattamento con paroxetina liquidando così la differenza clinicamente importante tra i gruppi paroxetina e placebo per gravi eventi avversi.
Effetti mascheranti di farmaci concomitanti
In quasi tutti gli studi, i pazienti assumeranno farmaci concomitanti. Gli eventi avversi di questi altri farmaci tenderanno a nascondere le differenze tra il trattamento farmacologico attivo e il placebo. Questo potrebbe essere un fattore importante nelle sperimentazioni di trattamenti come le statine, in cui i pazienti spesso assumono più farmaci. Di conseguenza, abbiamo anche confrontato l’incidenza di eventi avversi nei pazienti che assumevano farmaci concomitanti con l’incidenza in quelli che non assumevano altri farmaci. Altri farmaci sono stati istituiti nel corso dello studio che non abbiamo analizzato, ma i dati sono disponibili nelle tabelle K e L nell’appendice 2 e nei fogli di lavoro depositati su www.Study329.org e nell’appendice B dal CSR. Ci sono molti altri punti di vista nei dati disponibili su www.Study329.org che potrebbero essere ulteriormente esplorati, come gli effetti del ritiro di farmaci concomitanti sui profili degli eventi avversi, poiché i fogli di calcolo documentano il giorno di insorgenza degli eventi avversi e le date di inizio o interruzione di eventuali farmaci concomitanti. Un’altra opzione da esplorare è la possibilità di eventuali cascate di prescrizione innescate da eventi avversi correlati ai farmaci in studio.
Ignorare gli effetti della sospensione del farmaco
Il protocollo includeva una fase di scalaggio della durata di 7-17 giorni alla quale i ricercatori erano incoraggiati ad aderire, anche nei pazienti che avevano interrotto il trattamento a causa di eventi avversi. Il documento originale non ha analizzato questi dati separatamente. L’aumento dei tassi di eventi avversi psichiatrici emersi durante la fase di sospensione nella nostra analisi sono coerenti con la dipendenza e il ritiro dalla paroxetina, come riportato da Fava. 29
Processo RIAT
Questo esercizio RIAT si è rivelato estremamente impegnativo in termini di risorse. Abbiamo registrato oltre 250.000 parole di corrispondenza e-mail tra il team in due anni. L’interfaccia del desktop remoto a schermo singolo (che abbiamo chiamato “periscopio”) si è rivelata una sfida enorme. L’analisi dell’efficacia richiedeva che più tabelle di fogli di calcolo fossero aperte contemporaneamente, con molte operazioni di copia, incolla e controllo incrociato, e lo spazio era altamente restrittivo. L’accesso ai moduli dei rapporti sui casi richiedeva un’ampia corrispondenza con GSK. 12 Sebbene GSK alla fine fornisse i moduli di segnalazione dei casi, erano ancora più difficili da gestire, dato che potevamo vedere solo una pagina alla volta.
Ci sono volute circa mille ore per esaminare solo un terzo dei moduli dei rapporti sui casi. Non essere in grado di stamparli era un handicap considerevole. Non c’erano mezzi per preparare pacchetti per più codificatori indipendenti, per diminuire il bias; per fare annotazioni o usare commenti a margine; o per ordinare e raccogliere le segnalazioni di eventi avversi. La nostra esperienza evidenzia che le copie sia cartacee che elettroniche sono fondamentali per un’impresa come questa. La nostra analisi indica che sebbene le CSR siano utili, e in questo caso tutto ciò che era necessario per rianalizzare l’efficacia, l’analisi degli eventi avversi richiede l’accesso ai dati a livello di singolo paziente nei moduli di segnalazione dei casi. Poiché abbiamo aperto nuovi orizzonti, non abbiamo avuto precedenti a cui fare affidamento nell’analisi e nel reporting. Attendiamo con interesse altri sforzi per fare qualcosa di simile.
Punti di forza e limiti di questo studio
Lo studio 329 era uno studio controllato randomizzato con un campione di dimensioni ragionevoli. C’erano, tuttavia, prove di violazioni del protocollo, inclusi alcuni casi di rottura dell’accecamento. La codifica degli eventi avversi da parte degli investigatori originali ha sollevato la possibilità che alcuni altri dati potessero essere inaffidabili. Il processo è durato solo otto settimane. I partecipanti avevano una depressione relativamente cronica (durata media superiore a un anno), che limiterebbe la generalizzabilità dei risultati, in particolare nelle cure primarie, perché molti casi di depressione adolescenziale hanno durate più brevi. 28 La generalizzabilità all’assistenza primaria sarebbe inoltre limitata dal fatto che i partecipanti sono stati reclutati attraverso strutture terziarie.
L’analisi RIAT ha aperto nuovi orizzonti, ma è stata limitata in quanto è stato possibile controllare solo il 34% (93/275) dei moduli di segnalazione dei casi. Tempo e risorse hanno impedito l’accesso a tutti i moduli a causa delle difficoltà nell’utilizzo del portale per accedere ai dati dello studio e perché mancavano notevoli quantità di dati. L’analisi ha generato un’utile tassonomia dei potenziali ostacoli alla segnalazione accurata di eventi avversi e, anche tenendo conto delle limitazioni di cui sopra, ha mostrato il valore di consentire l’accesso ai dati.
Conclusione e implicazioni per la ricerca e la politica
Contrariamente al rapporto originale di Keller e colleghi, la nostra rianalisi dello studio 329 non ha mostrato alcun vantaggio di paroxetina o imipramina rispetto al placebo negli adolescenti con sintomi di depressione su nessuna delle variabili prespecificate. L’entità degli aumenti clinicamente significativi degli eventi avversi nei bracci paroxetina e imipramina, inclusi eventi avversi gravi, gravi e correlati al suicidio, è diventata evidente solo quando i dati sono stati resi disponibili per la rianalisi. I ricercatori e i medici dovrebbero riconoscere i potenziali pregiudizi nella ricerca pubblicata, comprese le potenziali barriere alla segnalazione accurata dei danni che abbiamo identificato. Le autorità di regolamentazione dovrebbero imporre l’accessibilità dei dati e dei protocolli. Come con la maggior parte degli articoli scientifici, Keller e colleghi danno l’impressione che “i dati abbiano parlato”. Questa posizione autorevole è possibile solo in assenza di accesso ai dati. Quando i dati diventano accessibili ad altri, diventa chiaro che la paternità scientifica è provvisoria piuttosto che autorevole.
Cosa si sa già su questo argomento
- Vi è una mancanza di accesso ai dati dalla maggior parte degli studi clinici controllati randomizzati, rendendo difficile rilevare segnalazioni di parte
- In assenza di accesso ai dati primari, le conclusioni fuorvianti nelle pubblicazioni di questi studi possono sembrare definitive
- Lo studio 329 di SmithKline Beecham, uno studio influente che ha riportato che la paroxetina era sicura ed efficace per gli adolescenti, è uno di questi studi
Cosa aggiunge questo studio
- Sulla base dell’accesso ai dati originali dello studio 329, riportiamo una nuova analisi che conclude che la paroxetina era inefficace e pericolosa in questo studio
- L’accesso ai dati primari rende evidenti i molti modi in cui i dati possono essere analizzati e rappresentati, mostrando l’importanza dell’accesso ai dati e il valore della rianalisi delle sperimentazioni
- Ci sono importanti implicazioni per la pratica clinica, la ricerca, la regolamentazione delle sperimentazioni, la concessione di licenze per i farmaci e la sociologia e la filosofia della scienza
- La nostra rianalisi ha richiesto lo sviluppo di metodi che potessero essere adattati per future rianalisi di studi randomizzati controllati
Note a piè di pagina
- Ringraziamo Carys Hogan per il lavoro sul database e Tom Jefferson e Leemon McHenry per i commenti sulle bozze precedenti.
- Lo studio SmithKline Beecham è stato registrato con il numero 29060/329. Il protocollo era lo studio SmithKline Beecham 29060/329, rapporto clinico finale (fase acuta), appendice a, protocollo, da p 531. 13 Lo studio è stato finanziato da SmithKline Beecham. Il protocollo di analisi dei dati per la rianalisi RIAT è stato presentato a GSK il 28 ottobre 2013 e approvato da GSK il 4 dicembre 2013.
- Collaboratori: Ideazione / progettazione dell’opera: DH, JJ, JMN. Acquisizione dei dati: JJ (negoziazione con GSK); CT e EA-J (RIATAR); JMN (dati di efficacia utilizzando il sistema remoto online GSK); JLN (danneggia i dati utilizzando il sistema remoto online GSK). Analisi dei dati: JMN (efficacia); JLN e DH (danni). Interpretazione dei dati: tutti gli autori. Redazione dell’opera e revisione critica per importanti contenuti intellettuali, approvazione finale della versione da pubblicare: tutti gli autori. Tutti gli autori accettano di essere responsabili di tutti gli aspetti del lavoro. JJ è il garante. I primi quattro autori hanno dato lo stesso contributo al documento.
- Finanziamento: questa ricerca non ha ricevuto alcuna sovvenzione specifica da alcuna agenzia di finanziamento nei settori pubblico, commerciale o no profit.
- Interessi in competizione: tutti gli autori hanno completato il modulo di divulgazione uniforme dell’ICMJE su www.icmje.org/coi_disclosure.pdfe dichiarare: DH è stata ed è un testimone esperto per i querelanti in casi legali che coinvolgono la paroxetina della droga di GlaxoSmithKline. È anche un testimone per i querelanti in azioni che coinvolgono altri antidepressivi con lo stesso meccanismo d’azione della paroxetina ed è nel comitato consultivo della Foundation for Excellence in Mental Health Care. DH e JLN sono membri fondatori di RxISK. JJ è stato pagato da Baum, Hedlund, Aristei e Goldman, Los Angeles, CA, per fornire analisi e opinioni di esperti sui documenti ottenuti da GlaxoSmithKline in una class action sullo Studio 329 e da Forest in relazione a studi clinici controllati randomizzati su citalopram pediatrici. Alcuni degli autori stanno discutendo con un editore accademico in merito all’adattamento del caso dello studio 329 come un libro per scopi educativi.
- Approvazione etica: il protocollo e la dichiarazione di consenso informato sono stati approvati da un comitato di revisione istituzionale prima dell’inizio di ciascun centro, in conformità con il 21 Codice dei regolamenti federali (CFR) parte 56 degli Stati Uniti. Il consenso informato scritto è stato ottenuto da ciascun paziente prima dell’ingresso nel studio, in conformità con 21 CFR Parte 50. Sono stati forniti moduli di case report per la registrazione dei dati di ciascun paziente (Final Clinical Report pag. 000030). Il consenso informato del campione è fornito nell’appendice del protocollo, appendice C, pp 000590-4. Non sono disponibili ulteriori informazioni in merito al particolare comitato di revisione istituzionale che ha approvato lo studio.
- Trasparenza: JJ afferma che il manoscritto è un resoconto onesto, accurato e trasparente dello studio riportato; che nessun aspetto importante dello studio è stato omesso; e che sono state spiegate eventuali discrepanze rispetto allo studio pianificato (e, se pertinente, registrato).
- Condivisione dei dati: rapporti di studi clinici, tabelle di dati dettagliate e codice di programmazione sono disponibili su Dryad Digital Repository ( http://dx.doi.org/10.5061/dryad.bv8j6 ) e su www.Study329.org/ .
Questo è un articolo ad accesso aperto distribuito in conformità con la licenza Creative Commons Attribution Non Commercial (CC BY-NC 4.0), che consente ad altri di distribuire, remixare, adattare, costruire su questo lavoro in modo non commerciale e concedere in licenza i loro lavori derivati su diversi termini, a condizione che l’opera originale sia correttamente citata e l’uso non sia commerciale. Vedi: http://creativecommons.org/licenses/by-nc/4.0/ .
Riferimenti bibiliografici
- Doshi P, Dickersin K, Healy D, Vedula SS, Jefferson T. Restoring invisible and abandoned trials: a call for people to publish the findings. BMJ 2013;346:f2865.
- Keller MB, Ryan ND, Strober M, et al. Efficacy of paroxetine in the treatment of adolescent major depression: a randomized, controlled trial. J Am Acad Child Adolesc Psychiatry 2001;40:762-72.
- McHenry L, Jureidini J. Industry-sponsored ghostwriting in clinical trial reporting: a case study. Account Res 2008;15:152-67.
- Jureidini J, McHenry L, Mansfield P. Clinical trials and drug promotion: selective reporting of study 329. Int J Risk Saf Med 2008;20:73-81.
- Jureidini J, McHenry L. Conflicted medical journals and the failure of trust. Account Res 2011;18:45-54.
- Kraus JE, letter to Jon Jureidini. 2013. www.bmj.com/content/ suppl/2013/11/12/bmj.f6754.DC1/doshinov16.ww1_default.pdf.
- Treasure T, Monson K, Fiorentino F, Russell C. The CEA Second-Look Trial: a randomised controlled trial of carcinoembryonic antigen prompted reoperation for recurrent colorectal cancer. BMJ Open 2014 May 13;4:e004385.
- Ebrahim S, Sohani ZN, Montoya L, et al. Reanalyses of randomized clinical trial data. JAMA 2014;312:1024-32.
- SmithKline Beecham. A multi-center, double-blind, placebo controlled study of paroxetine and imipramine in adolescents with unipolar major depression –acute phase, Final clinical report. www.gsk.com/ media/389566/depression_329_full.pdf.
- Healthy Skepticism International News. Paxil Study 329: paroxetine vs imipramine vs placebo in adolescents. 2010. www.healthyskepticism. org/global/news/int/hsin2010-01.
- SAS Solutions OnDemand. www.ondemand.sas.com/sam/? sid=1393012805&rid=DqqHX0rCqAWZ7TJlCIPijRTscQ.
- Correspondence between Jureidini and GSK. Rapid responses to putting GlaxoSmithKline to the test over paroxetine. BMJ 2013;347:f6754. www. bmj.com/content/347/bmj.f6754/rapid-responses
- SmithKline Beecham. A multi-center, double-blind, placebo controlled study of paroxetine and imipramine in adolescents with unipolar major depression 1993/amended 1996. www.gsk.com/media/ 360485/329-AppA.PDF.
- Diagnostic and statistical manual of mental disorders, third edition, revised (DSM-III-R). American Psychiatric Association, 1987.
- Fawcett J, Epstein P, Fiester SJ, Elkin I, Autry JH. Clinical management— imipramine/placebo administration manual. NIMH Treatment of Depression Collaborative Research Program. Psychopharmacol Bull 1987;23:309-24.
- Hamilton M. Development of a rating scale for primary depressive illness. Br J Soc Clin Psychol 1967;6:278-96.
- Sigafoos AD, Feinstein CB, Damond M, Reiss D. The measurement of behavioral autonomy in adolescence: the Autonomous Functioning Checklist. Adolesc Psychiatry 1988;15:432-62.
- SKB. Draft Minutes: 4/22/97 Teleconference. Paroxetine Study 329 efficacy analysis. www.healthyskepticism.org/files/docs/gsk/ paroxetine/study329/970422teleconference.pdf.
- GlaxoSmithKline, Paroxetine—paediatric and adolescent patients. www.gsk.com/en-gb/media/resource-centre/paroxetine/ paroxetine-paediatric-and-adolescent-patients/.
- Winter C. MedDRA in clinical trials—industry perspective SFDA‐ICH MedDRA Workshop, Beijing, 13‐14 May 2011. www.meddra.org/ sites/default/files/page/documents_insert/christina_winter_2_ meddra_in_clinical_trials_industry_perspective.pdf.
- Jureidini JN, Nardo JM. Inadequacy of remote desktop interface for independent reanalysis of data from drug trials. BMJ 2014;349:g4353.
- Fitzgerald K, Healy D. Dystonias and dyskinesias of the jaw associated with the use of SSRIs. Human Psychopharmacol 1995;10:215-20.
- Kline RB. Beyond significance testing. Statistics reform in the behavioral sciences. 2nd ed. American Psychological Association, 2013.
- R Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing. www.R-project.org/.
- Brecher M. Review and evaluation of clinical data. Original NDA 20—031. Paroxetine (Aropax). Efficacy review. SmithKline Beecham Pharmaceuticals, 1991.
- Hughes S, Cohen, D, Jaggi R. Differences in reporting serious adverse events in industry sponsored clinical trial registries and journal articles on antidepressant and antipsychotic drugs: a cross-sectional study. BMJ Open 2014;4:e005535.
- Maund E, Tendal B, Hróbjartsson A, et al. Benefits and harms in clinical trials of duloxetine for treatment of major depressive disorder: comparison of clinical study reports, trial registries, and publications. BMJ 2014;348:g3510
- Lewinsohn PM, Clarke GN, Seeley JR, Rohde P. Major depression in community adolescents: age at onset, episode duration, and time to recurrence. J Am Acad Child Adolesc Psychiatry 1994;33:809-18.
- Fava M. Prospective studies of adverse events related to antidepressant discontinuation. J Clin Psychiatry 2006;67(suppl 4):14-21.
